
15221734409



来来来,朋友们,今天给大家分享个厉害的杂志~
先听听它的名头:心血管领域的期刊、创刊久且影响力持续在线,地位很高啊,它就是 Circulation!
影响因子:多年保持在较高水平,如 2024 年 IF 为35.5,一直稳居心血管领域前列。
SSN:0009-7322(P-ISSN)。
分区:中科院医学 1 区 TOP,小类 “心脏和心血管系统” 及 “外周血管病” 均为 1 区,JCR 分区为 Q1,排名靠前。
发文量:2024约 245 篇。
发刊频率:每周
审稿速度:
原创研究文章的交付时间:从投稿到次决定:17天;接受提前印刷出版:7-10天;接受印刷出版:81天。
版面费:
CIRCULATION是一本混合OA期刊。如果该刊采用的是AHA许可协议—CC-BY-NC-ND,文章发表费为3806美元(约人民币27843)。如果该刊采用的是AHA许可协议—CC-BY,则需要文章发表费5057美元(约人民币36996)。
这个杂志的审稿标准严格,论文质量高,心血管相关的基础研究、临床研究等各类文章在上面都有发表,大家如果从事心血管领域相关研究,投稿冲击一波很值得!
背景:
心脏衰竭作为各种心血管疾病的终末阶段,已被公认为全球死亡率和发病率的主要原因。从适应性心脏肥大到适应不良心脏肥大的转变与心力衰竭的进展密切相关,是心脏死亡率的主要危险因素。尽管目前投入了精力来研究适应不良的心脏肥大,但心脏肥大的潜在机制尚不清楚。
近的研究强调了细胞器相互作用在维持细胞稳态和调节各种病理过程(包括病理性心脏肥大)中的关键作用。线粒体相关ER膜(MAM)是由特异性栓系和间隔蛋白形成的动态微结构域,在Ca2+信号传导、脂质转运和代谢中起着至关重要的作用。MAMs 在从适应性心肌肥大过渡到适应不良的心肌肥大期间发生改变,导致心力衰竭进展。然而,MAM结构和功能如何受到调节并导致心力衰竭的进展(即病理性心脏肥大)仍未阐明。
在本项研究中,测序揭示了 MAMs 中一关键因子的变化,并通过进一步的实验验证,确定了一种MAMs 复合物的新成分,而且为病理性心脏肥大提供了新的治疗靶点。
结论1: FMO2在心肌肥厚中表达下调并定位于MAM结构
研究团队通过测序发现 485 个差异表达基因,涉及内质网膜及 MAM 结构相关通路,FMO2 作为关键下调分子被识别。经实验验证,心肌肥厚患者样本中 FMO2 低表达。在 TAC 手术小鼠模型及多种动物和体外模型中也观察到 FMO2 下调,表明其在心肌肥厚中广泛存在且高度一致,可能在心肌重构中扮演关键角色。
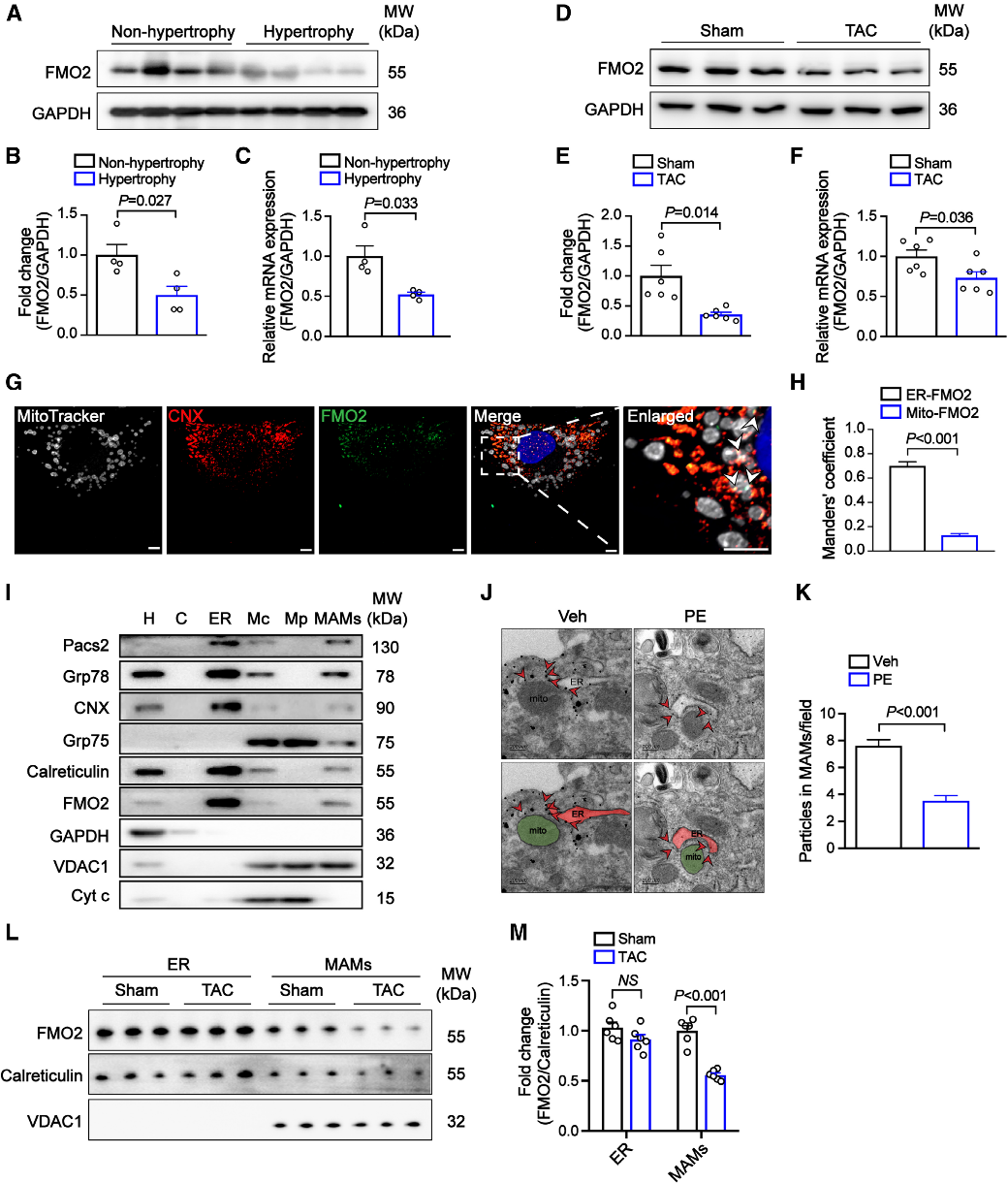
Fig1. FMO2在心肌肥厚中表达下调并定位于MAM结构
结论2: FMO2心肌特异性缺失加重心肌肥厚
研究团队通过构建全身敲除(FMO2⁻/⁻)和心肌特异性敲除(FMO2^cko)小鼠模型,并利用TAC诱导心肌肥厚,探究FMO2的心脏保护功能。进一步利用Myh6-Cre/ERT2系统构建的FMO2^cko小鼠在TAC后也呈现类似的病理改变。实验结果表明,FMO2在心肌细胞中具有抗心肌肥厚的保护作用,其缺失会加重病理性心肌重构进程。
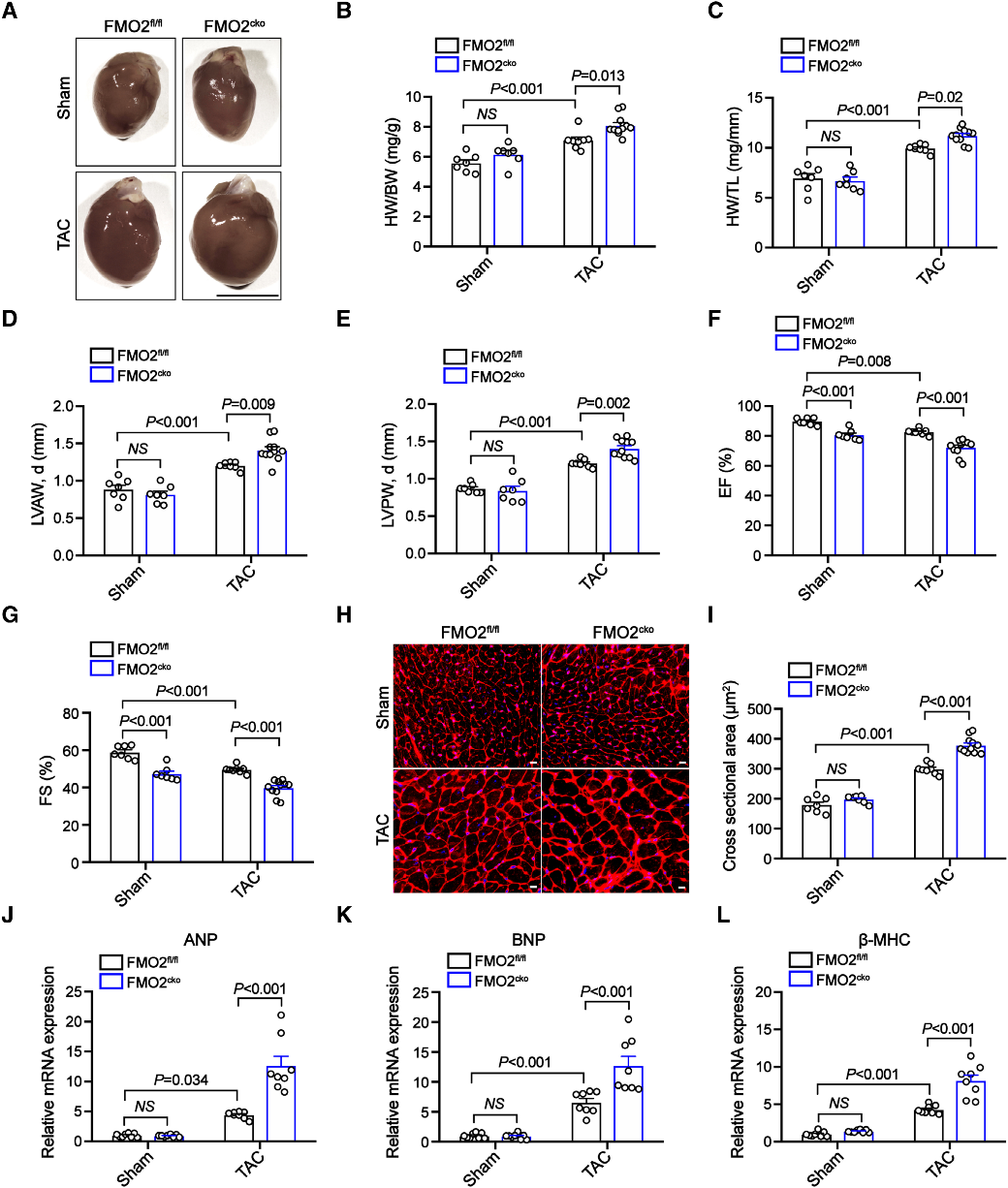
Fig2. FMO2心肌特异性缺失加重心肌肥厚
结论3:特异性心脏过表达FMO2可减轻心脏肥大
研究者利用AAV9-cTnT系统在小鼠心脏特异性过表达FMO2,4周后 实验结果确认表达升高。对小鼠实施TAC手术后,FMO2过表达组心重比降低,组织染色观察到心肌细胞横截面积较小,肥厚相关基因表达被抑制。结果表明,FMO2过表达可缓解心肌结构重构,有效保护心功能,具有抗心肌肥厚作用。
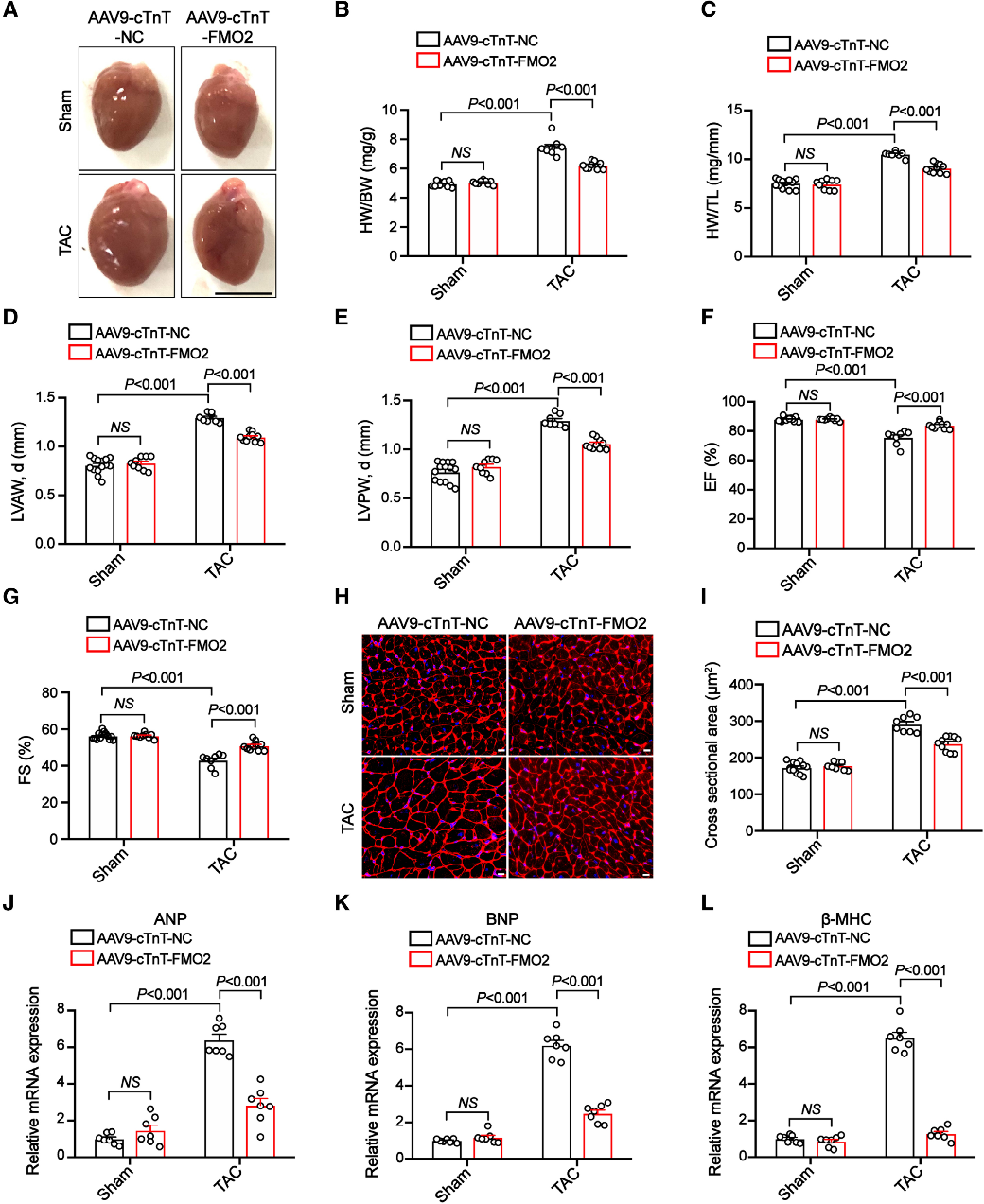
Fig3. 特异性心脏过表达FMO2可减轻心脏肥大
结论4:FMO2 在心脏肥大期间保持 ER-线粒体接近度
在苯肾上腺素刺激的NRCMs中,操控FMO2表达显示,FMO2可调控ER与线粒体共定位,敲低加剧解耦合,过表达恢复接触。电子显微镜证实FMO2缺失削弱MAM接触面积,过表达可恢复,且MAM类型不变,表明其通过非传统连接蛋白途径调控结构。FMO2敲低影响线粒体形态结构,过表达逆转改变。这些结果在TAC模型中得到验证,表明FMO2通过维持ER-线粒体耦联状态,在防止病理性心肌肥厚中发挥关键作用,且机制独立于传统MAM连接蛋白。
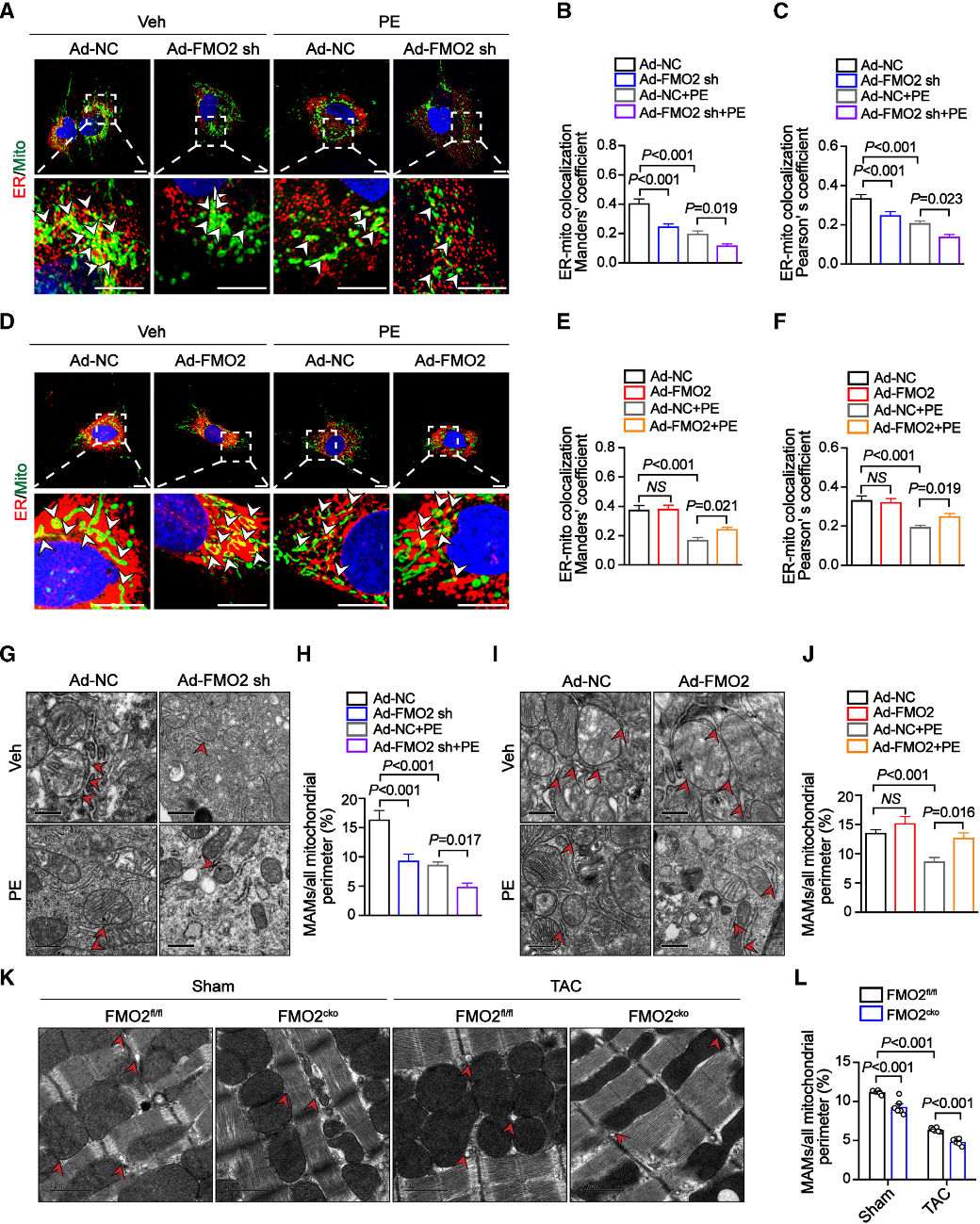
Fig4. FMO2 在心脏肥大期间保持 ER-线粒体接近度
结论5:FMO2 通过与 IP3R2-GRP75-VDAC1 复合物相互作用参与 MAMs 的形成
研究发现,FMO2缺失显著降低线粒体与内质网组分的结合能力,表明其参与 MAM 结构组装。FMO2 敲低削弱 IP3R2 对 Grp75 结合力,FMO2⁻/⁻ 小鼠 MAM 组分中 IP3R2 水平下降。PLA 实验验证 FMO2 与三元复合物空间接近性,且苯肾上腺素处理或 TAC 手术致 PLA 信号下降,FMO2 过表达可恢复,缺失则加重信号丢失。研究表明,FMO2 通过与 IP3R2–Grp75–VDAC1 复合物相互作用稳定 MAM 结构,是其形成和维持的关键调控因子。
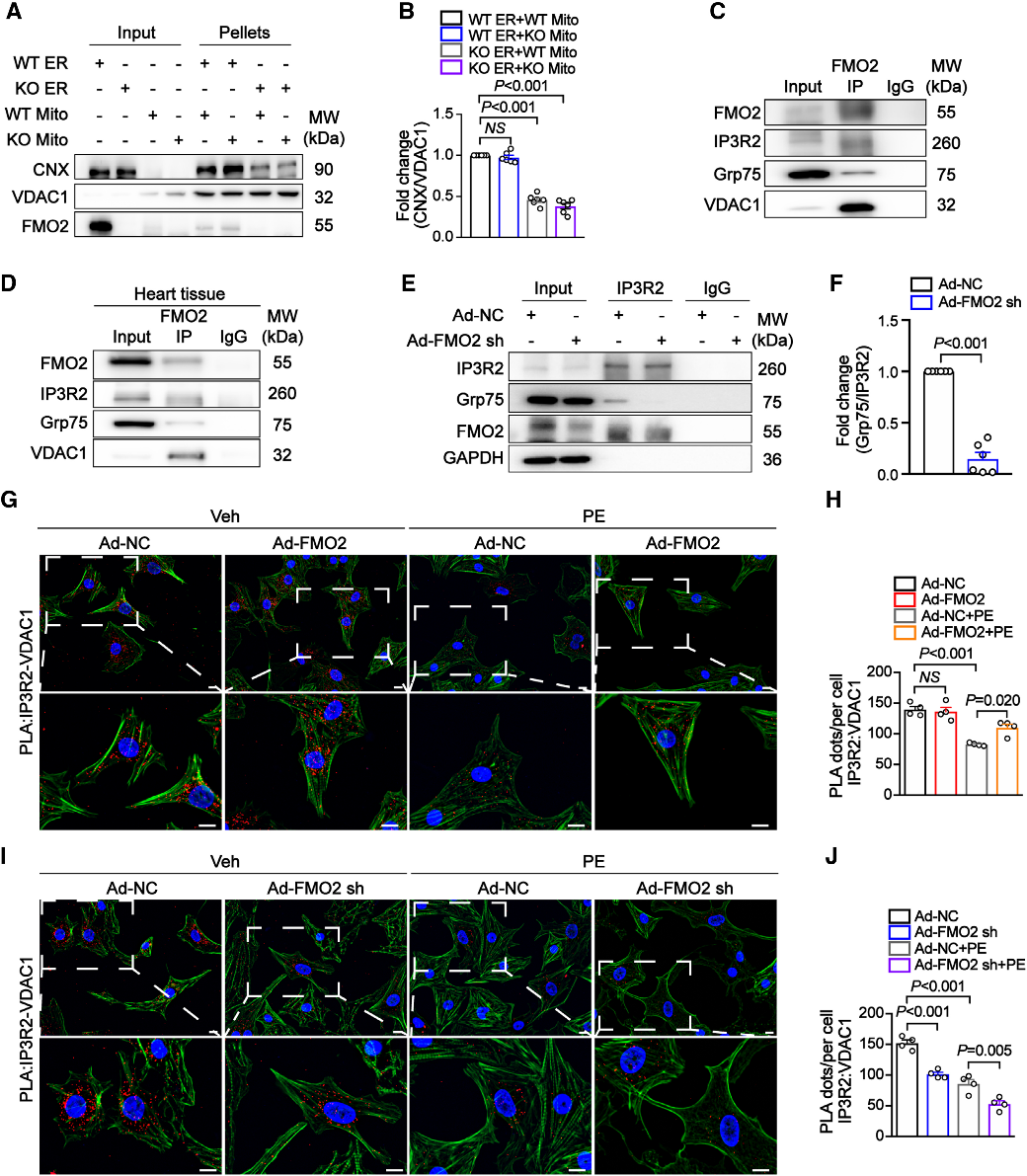
Fig5. FMO2 通过与 IP3R2-GRP75-VDAC1 复合物相互作用参与 MAMs 的形成
结论6:MAM 定位的 FMO2 保护 NRCM 免受肥大
为验证FMO2抗心肌肥大作用是否依赖其MAMs定位,研究构建了缺失内质网跨膜区的FMO2突变体Fdel510-535并稳定表达于新生鼠心肌细胞。结果显示,其无法恢复Ca²⁺转运,不能增强蛋白互作信号或修复解偶联,也不能改善细胞肥大及基因表达。但酶活缺失型FMO2突变体Ad-FMO2 mut可恢复Ca²⁺转运、改善线粒体–内质网结构、抑制肥大及标志物上调。表明FMO2通过MAM定位而非酶活性发挥抗心肌肥大作用。
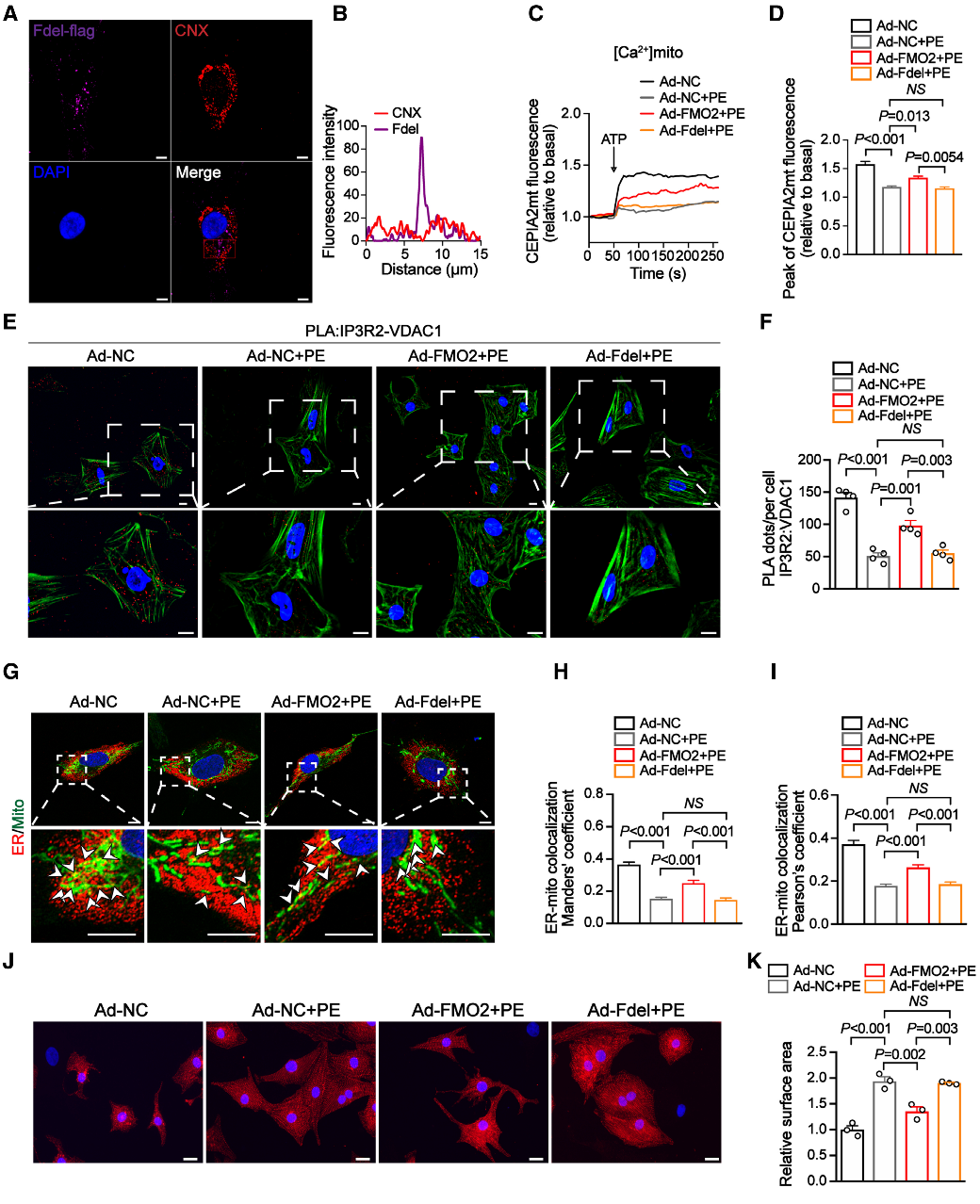
Fig6. MAM 定位的 FMO2 保护 NRCM 免受肥大
结论7:合成接头诱导的 ER-线粒体接触可防止心脏肥大
研究发现FMO2缺失致心肌肥大与内质网–线粒体接触受损相关。在TAC小鼠模型中,该连接蛋白改善心脏结构和功能,抑制病理性重构,增强线粒体功能,缓解FMO2⁻/⁻小鼠的心肌肥大,表明增强ER–线粒体连接可抵抗FMO2缺失引发的心肌病理性改变。
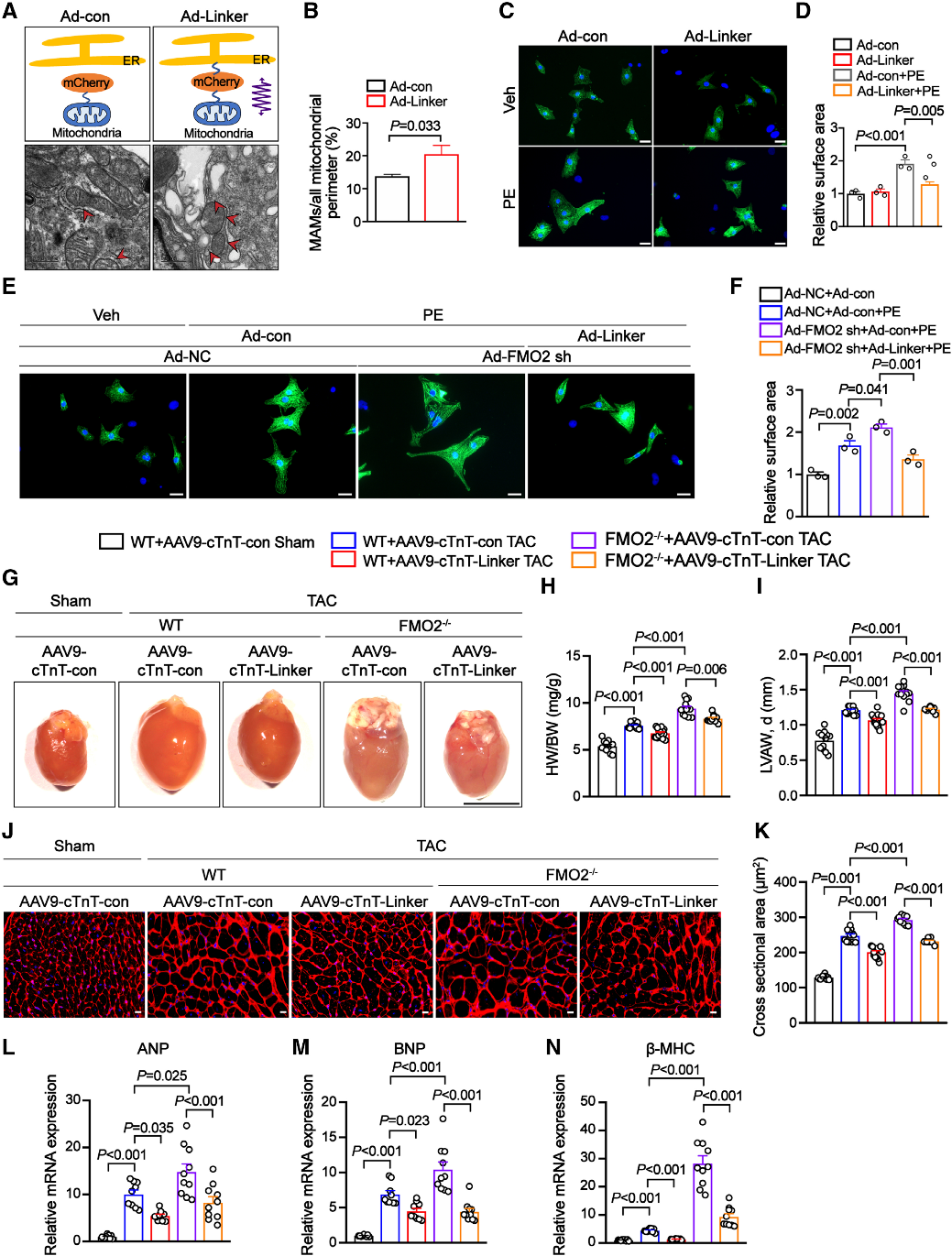
Fig7. 合成接头诱导的 ER-线粒体接触可防止心脏肥大
研究亮点
· 揭示FMO2在心脏中通过维持MAM结构调控心肌肥厚;
· 连接基础研究与转化医学:提出以“ER-线粒体接触增强”为潜在干预策略;
· 临床前数据充分,包括人类心脏组织样本、小鼠模型、干细胞心肌细胞等多个体系验证。
 欢迎来到
欢迎来到
